
Chromosomes
What is a chromosome?
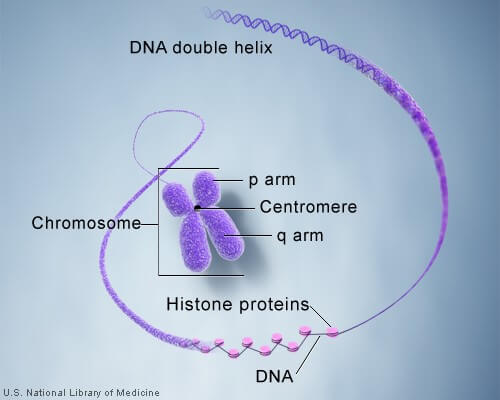
In the nucleus of each cell, the DNA molecule is packaged into thread-like structures called chromosomes. Each chromosome is made up of DNA tightly coiled many times around proteins called histones that support its structure.
Chromosomes are not visible in the cell’s nucleus—not even under a microscope—when the cell is not dividing. However, the DNA that makes up chromosomes becomes more tightly packed during cell division and is then visible under a microscope. Most of what researchers know about chromosomes was learned by observing chromosomes during cell division.
Each chromosome has a constriction point called the centromere, which divides the chromosome into two sections, or “arms.” The short arm of the chromosome is labeled the “p arm.” The long arm of the chromosome is labeled the “q arm.” The location of the centromere on each chromosome gives the chromosome its characteristic shape, and can be used to help describe the location of specific genes.
DNA and histone proteins are packaged into structures called chromosomes.

Chromosome 1
There are 890 known diseases related to chromosome 1.
Some of these diseases are deafness, Alzheimer disease, glaucoma and breast cancer. Rearrangements and mutations of chromosome 1 are prevalent in cancer and many other diseases. Patterns of sequence variation reveal signals of recent selection in specific genes that may contribute to human fitness, and also in regions where no function is evident. The following diseases are some of those related to genes on chromosome 1 (which contains the most known genetic diseases of any human chromosome):
Brooke GreenbergDisease (Syndrome X)
Carnitine palmitoyltransferase II deficiency
Charcot–Marie–Tooth disease, types 1 and 2
collagenopathy, types II and XI
Deafness, autosomal recessive deafness 36
Ehlers-Danlos syndrome, kyphoscoliosis type
Familial adenomatous polyposis
Gelatinous drop-like corneal dystrophy
Hepatoerythropoietic porphyria
Hutchinson Gilford Progeria Syndrome
3-hydroxy-3-methylglutaryl-CoA lyase deficiency
Hypertrophic cardiomyopathy, autosomal dominant mutations of TNNT2; hypertrophy usually mild; restrictive phenotype may be present; may carry high risk of sudden cardiac death
medium-chain acyl-coenzyme A dehydrogenase deficiency
Nonsyndromic deafness, autosomal dominant
Chromosome 2
The following diseases and traits are related to genes located on chromosome 2:
2p15-16.1 microdeletion syndrome
Amyotrophic lateral sclerosis, type 2
Ehlers–Danlos syndrome, classical type
Ehlers–Danlos syndrome, vascular type
Fibrodysplasia ossificans progressiva
Hereditary nonpolyposis colorectal cancer
Infantile-onset ascending hereditary spastic paralysis
Juvenile primary lateral sclerosis
Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency
Maturity onset diabetes of the youngtype 6
Mitochondrial trifunctional protein deficiency
Nonsyndromic deafness, autosomal recessive
Primary pulmonary hypertension
Sitosterolemia(knockout of either ABCG5 or ABCG8)
Chromosome 3
The following diseases are some of those related to genes on chromosome 3:
3-methylcrotonyl-CoA carboxylase deficiency
Arrhythmogenic right ventricular dysplasia
Blepharophimosis, epicanthus inversus and ptosis type 1
Breast/colon/lung/pancreatic cancer
Carnitine-acylcarnitine translocase deficiency
Cerebral cavernous malformation
Charcot-Marie-Tooth disease, type 2
Chromosome 3q duplication syndrome
Dystrophic epidermolysis bullosa
Endplate acetlycholinesterase deficiency
Heart block, progressive/nonprogressive
Hereditary nonpolyposis colorectal cancer
HIVinfection, susceptibility/resistance to
Hypobetalipoproteinemia, familial
Leukoencephalopathy with vanishing white matter
Malignant hyperthermia susceptibility
Metaphyseal chondrodysplasia, Murk Jansen type
Muir-Torre family cancer syndrome
Neuropathy, hereditary motor and sensory, Okinawa type
Nonsyndromic deafness, autosomal recessive
Chromosome 4
The following are some of the diseases related to genes located on chromosome 4:
autosomal dominant polycystic kidneydisease (PKD-2)
Crouzonodermoskeletal syndrome
Facioscapulohumeral muscular dystrophy
Fibrodysplasia ossificans progessiva FOP
nonsyndromic deafness, autosomal dominant
tetrahydrobiopterin deficiency
thanatophoric dysplasia, type 1
Chromosome 5
The following are some of the diseases related to genes located on chromosome 5:
Corneal dystrophy of Bowman layer, type I
Corneal dystrophy of Bowman layer, type II
Ehlers-Danlos syndrome, dermatosparaxis type
Familial adenomatous polyposis
Granular corneal dystrophy type I
Granular corneal dystrophy type II
GM2-gangliosidosis, AB variant
3-Methylcrotonyl-CoA carboxylase deficiency
Recessive multiple epiphyseal dysplasia
Survival motor neuron spinal muscular atrophy
Tricho-hepato-enteric syndrome
The following conditions are caused by changes in the structure or number of copies of chromosome 5:
Cri-du-chat syndrome is caused by a deletion of the end of the short (p) arm of chromosome 5. This chromosomal change is written as 5p-. The signs and symptoms of cri-du-chat syndrome are probably related to the loss of multiple genes in this region. Researchers have not identified all of these genes or determined how their loss leads to the features of the disorder. They have discovered, however, that a larger deletion tends to result in more severe mental retardation and developmental delays in people with cri-du-chat syndrome.
Researchers have defined narrow regions of the short arm of chromosome 5 that are associated with particular features of cri-du-chat syndrome. A specific region designated 5p15.3 is associated with a cat-like cry, and a nearby region called 5p15.2 is associated with mental retardation, small head (microcephaly), and distinctive facial features.
Familial Adenomatous Polyposis is caused by a deletion of the APC tumor suppressor gene on the long (q) arm of chromosome 5. This chromosomal change results in thousands of colonic polyps which gives the patient a 100% risk of colon cancer if total colectomy is not done.
Chromosome 5q deletion syndrome is caused by the deletion of the q arm (long arm) of chromosome 5. This deletion has been linked to several blood related disorders including Myelodysplastic syndrome andErythroblastopenia. This is a different condition then Cri-du-chat which was mentioned above.
Other changes in the number or structure of chromosome 5 can have a variety of effects, including delayed growth and development, distinctive facial features, birth defects, and other medical problems. Changes to chromosome 5 include an extra segment of the short (p) or long (q) arm of the chromosome in each cell (partial trisomy 5p or 5q), a missing segment of the long arm of the chromosome in each cell (partial monosomy 5q), and a circular structure called ring chromosome A ring chromosome occurs when both ends of a broken chromosome are reunited.
Chromosome 6
The following diseases are some of those related to genes on chromosome 6:
ankylosing spondylitis, HLA-B
collagenopathy, types II and XI
Coeliac diseaseHLA-DQA1 & DQB1
Ehlers-Danlos syndrome, classical, hypermobility, and Tenascin-X types
Autosomal nonsyndromic deafness
otospondylomegaepiphyseal dysplasia
Rheumatoid arthritis, HLA-DR
Diabetes mellitus type 1, HLA-DR, DQA1 & DQB1
Chromosome 7
The following diseases are some of those related to genes on chromosome 7:
cerebral cavernous malformation
Charcot–Marie–Tooth disease, type 2
congenital bilateral absence of vas deferens
distal spinal muscular atrophy, type V
Ehlers–Danlos syndrome, arthrochalasia type
Ehlers–Danlos syndrome, classical type
hereditary nonpolyposis colorectal cancer
maturity onset diabetes of the youngtype 3
mucopolysaccharidosis type VIIor Sly syndrome
nonsyndromic deafness, autosomal dominant
nonsyndromic deafness, autosomal recessive
osteogenesis imperfecta, type I
osteogenesis imperfecta, type III
osteogenesis imperfecta, type II
osteogenesis imperfecta, type IV
p47-phox-deficient chronic granulomatous disease
Tritanopia or tritanomaly color blindness
The following conditions are caused by changes in the structure or number of copies of chromosome 7:
Williams syndromE is caused by the deletion of genetic material from a portion of the long (q) arm of chromosome 7. The deleted region, which is located at position 11.23 (written as 7q11.23), is designated as the Williams syndrome critical region. This region includes more than 20 genes, and researchers believe that the characteristic features of Williams syndrome are probably related to the loss of multiple genes in this region.
While a few of the specific genes related to Williams syndrome have been identified, the relationship between most of the genes in the deleted region and the signs and symptoms of Williams syndrome is unknown.
Other changes in the number or structure of chromosome 7 can cause delayed growth and development, mental disorder, characteristic facial features, skeletal abnormalities, delayed speech, and other medical problems. These changes include an extra copy of part of chromosome 7 in each cell (partial trisomy7) or a missing segment of the chromosome in each cell (partial monosomy 7). In some cases, several DNA building blocks (nucleotides) are deleted or duplicated in part of chromosome 7. A circular structure called ring chromosome 7 is also possible. A ring chromosome occurs when both ends of a broken chromosome are reunited.
Chromosome 8
The following diseases are some of those related to genes on chromosome 8:
Charcot-Marie-Tooth disease, type 2
Charcot-Marie-Tooth disease, type 4
Lipoprotein lipase deficiency, familial
Primary microcephaly
Rothmund-Thomson syndrome, or poikiloderma congenitale
Schizophrenia, associated with 8p21-22 locus
Chromosome 9
The following diseases are some of those related to genes on chromosome 9:
Chronic myelogenous leukemia(t9;22 – the Philadelphia chromosome)
Ehlers-Danlos syndrome, classical type
Gorlin syndromeor Nevoid Basal Cell Carcinoma syndrome
hereditary hemorrhagic telangiectasia
lethal congenital contracture syndrome
nonsyndromic deafness, autosomal dominant
nonsyndromic deafness, autosomal recessive
Chromosome 10
The following diseases are some of those related to genes on chromosome 10:
Beare-Stevenson cutis gyrata syndrome
Charcot-Marie-Tooth disease, type 1
Charcot-Marie-Tooth disease, type 4
congenital erythropoietic porphyria
multiple endocrine neoplasia type 2
nonsyndromic deafness, autosomal recessive
tetrahydrobiopterin deficiency
Chromosome 11
The following diseases are some of those related to genes on chromosome 11:
carnitine palmitoyltransferase I deficiency
Charcot-Marie-Tooth disease, type 4
Hereditary angioedemaOMIM: 106100
Jervell and Lange-Nielsen syndrome
Mantle cell lymphoma(t11;14)
methemoglobinemia, beta-globin type
multiple endocrine neoplasia type 1
nonsyndromic deafness, autosomal dominant
nonsyndromic deafness, autosomal recessive
Chromosome 12
The following diseases are some of those related to genes on chromosome 12:
collagenopathy, types II and XI
hereditary hemorrhagic telangiectasia
maturity onset diabetes of the youngtype 3
nonsyndromic deafness, autosomal dominant
Pallister-Killian syndrome(tetrasomy 12p)
spondyloepimetaphyseal dysplasia, Strudwick type
spondyloepiphyseal dysplasia congenita
Chromosome 13
The following diseases are some of those related to genes on chromosome 13:
Maturity onset diabetes of the youngtype 4
Nonsyndromic deafness, autosomal dominant
Nonsyndromic deafness, autosomal recessive
Chronic Lymphocytic Leukemia(Acquired defect)
The following conditions are caused by changes in the structure or number of copies of chromosome 13:
Retinoblastoma: A small percentage of retinoblastoma cases are caused by deletions in the region of chromosome 13 (13q14) containing the RB1 gene. Children with these chromosomal deletions may also have mental retardation, slow growth, and characteristic facial features (such as prominent eyebrows, a broad nasal bridge, a short nose, and ear abnormalities). Researchers have not determined which other genes are located in the deleted region, but a loss of several genes is likely responsible for these developmental problems.
Trisomy 13: Trisomy 13 occurs when each cell in the body has three copies of chromosome 13 instead of the usual two copies. Trisomy 13 can also result from an extra copy of chromosome 13 in only some of the body’s cells (mosaic trisomy 13). In a small percentage of cases, trisomy 13 is caused by a rearrangement of chromosomal material between chromosome 13 and another chromosome. As a result, a person has the two usual copies of chromosome 13, plus extra material from chromosome 13 attached to another chromosome. These cases are called translocation trisomy 13. Extra material from chromosome 13 disrupts the course of normal development, causing the characteristic signs and symptoms of trisomy 13. Researchers are not yet certain how this extra genetic material leads to the features of the disorder, which include severely abnormal cerebral functions, a small cranium, retardation, non functional eyes and heart defects.
Other chromosomal conditions: Partial monosomy 13qis a rare chromosomal disorder that results when a piece of the long arm (q) of chromosome 13 is missing (monosomic). Infants born with partial monosomy 13q may exhibit low birth weight, malformations of the head and face (craniofacial region), skeletal abnormalities (especially of the hands and feet), and other physical abnormalities. Mental retardation is characteristic of this condition. The mortality rate during infancy is high among individuals born with this disorder. Almost all cases of partial monosomy 13q occur randomly for no apparent reason (sporadic).
Chromosome 14
The following diseases are some of those related to genes on chromosome 14:
alpha-1 antitrypsin deficiency
Burkitt’s lymphoma(t8;14)
Follicular lymphoma(t14;18)
nonsyndromic deafness, autosomal dominant
tetrahydrobiopterin deficiency
Uniparental disomy(UPD) 14
Chromosome 15
The following diseases are some of those related to genes on chromosome 15.
The following conditions are caused by mutations in chromosome 15.
Two of the conditions (Angelman syndrome and Prader-Willi syndrome) involve a loss of gene activity in the same part of chromosome 15, the 15q11-q13 region. This discovery provided the first evidence in humans that something beyond genes could determine how the genes are expressed.
Angelman syndrome
Main article: Angelman syndrome
The main characteristics of Angelman syndrome are severe mental retardation, ataxia, lack of speech, and excessively happy demeanor. Angelman syndrome results from a loss of gene activity in a specific part of chromosome 15, the 15q11-q13 region. This region contains a gene called UBE3A that, when mutated or absent, likely causes the characteristic features of this condition. People normally have two copies of the UBE3A gene, one from each parent. Both copies of this gene are active in many of the body’s tissues. In the brain, however, only the copy inherited from a person’s mother (the maternal copy) is active. If the maternal copy is lost because of a chromosomal change or a gene mutation, a person will have no working copies of the UBE3A gene in the brain.
In most cases (about 70%), people with Angelman syndrome have a deletion in the maternal copy of chromosome 15. This chromosomal change deletes the region of chromosome 15 that includes the UBE3A gene. Because the copy of the UBE3A gene inherited from a person’s father (the paternal copy) is normally inactive in the brain, a deletion in the maternal chromosome 15 results in no active copies of the UBE3A gene in the brain.
In 3% to 7% of cases, Angelman syndrome occurs when a person has two copies of the paternal chromosome 15 instead of one copy from each parent. This phenomenon is called paternal uniparental disomy (UPD). People with paternal UPD for chromosome 15 have two copies of the UBE3A gene, but they are both inherited from the father and are therefore inactive in the brain.
About 10% of Angelman syndrome cases are caused by a mutation in the UBE3A gene, and another 3% result from a defect in the DNA region that controls the activation of the UBE3A gene and other genes on the maternal copy of chromosome 15. In a small percentage of cases, Angelman syndrome may be caused by a chromosomal rearrangement called a translocation or by a mutation in a gene other than UBE3A. These genetic changes can abnormally inactivate the UBE3A gene.
Angelman syndrome can be hereditary, as evidenced by one case where a patient became pregnant with a daughter who also had the condition.
Prader-Willi syndrome
Main article: Prader-Willi syndrome
The main characteristics of this condition include polyphagia (extreme, unsatiable appetite), mild to moderate developmental Delay, hypogonadism resulting in delayed to no puberty, and hypotonia. Prader-Willi syndrome is caused by the loss of active genes in a specific part of chromosome 15, the 15q11-q13 region. People normally have two copies of this chromosome in each cell, one copy from each parent. Prader-Willi syndrome occurs when the paternal copy is partly or entirely missing.
In about 70% of cases, Prader-Willi syndrome occurs when the 15q11-q13 region of the paternal chromosome 15 is deleted. The genes in this region are normally active on the paternal copy of the chromosome and are inactive on the maternal copy. Therefore, a person with a deletion in the paternal chromosome 15 will have no active genes in this region.
In about 25% of cases, a person with Prader-Willi syndrome has two maternal copies of chromosome 15 in each cell instead of one copy from each parent. This phenomenon is called maternal uniparental disomy. Because some genes are normally active only on the paternal copy of this chromosome, a person with two maternal copies of chromosome 15 will have no active copies of these genes.
In a small percentage of cases, Prader-Willi syndrome is not caused by a chromosomal rearrangement called a trans location. Rarely, the condition is caused by an abnormality in the DNA region that controls the activity of genes on the paternal chromosome 15. Because patients almost always have difficulty reproducing, Prader-Willi syndrome is generally not hereditary.
Isodicentric chromosome 15
A specific chromosomal change called an isodicentric chromosome 15 (previously called an inverted duplication 15) can affect growth and development. The patient possesses an “extra” or “marker” chromosome. This small extra chromosome is made up of genetic material from chromosome 15 that has been abnormally duplicated (copied) and attached end-to-end. In some cases, the extra chromosome is very small and has no effect on a person’s health. A larger isodicentric chromosome 15 can result in weak muscle tone (hypotonia), mental retardation, seizures, and behavioral problems. Signs and symptoms of autism (a developmental disorder that affects communication and social interaction) have also been associated with the presence of an isodicentric chromosome 15.
Chromosome 16
The following diseases are some of those related to genes on chromosome 16:
Familial Mediterranean fever(FMF)
Autosomal dominant polycystic kidneydisease (PKD-1)
Chromosome 17
The following diseases are related to genes on chromosome 17:
Cerebroretinal microangiopathy with calcifications and cysts
Charcot-Marie-Tooth disease, type 1
Ehlers-Danlos syndrome, arthrochalasia type
Ehlers-Danlos syndrome, classical type
Epidermodysplasia verruciformis
Glycogen storage disease type II(Pompe disease)
Hereditary neuropathy with liability to pressure palsies
Maturity onset diabetes of the youngtype 5
Nonsyndromic deafness, autosomal dominant
Nonsyndromic deafness, autosomal recessive
Osteogenesis Imperfecta, Type I
Osteogenesis Imperfecta, Type II
Osteogenesis Imperfecta, Type III
Chromosome 18
The following diseases are some of those related to genes on chromosome 18:
Chromosome 19
The following diseases are some of those related to genes on chromosome 19:
Alternating hemiplegia of childhood
Centronuclear myopathyautosomal dominant form
[(Congenital and inherited deafness)]
Myotubular myopathyautosomal dominant form
X-linked agammaglobulinemiaor Bruton’s Disease
Chromosome 20
The following diseases are some of those related to genes on chromosome 20:
Albright’s hereditary osteodystrophy
Adenosine deaminase deficiency
Maturity onset diabetes of the youngtype 1
Neuronal ceroid lipofuscinosis
Pantothenate kinase-associated neurodegeneration
Transmissible spongiform encephalopathy(prion diseases)
Chromosome 21
The following diseases are some of those related to genes on chromosome 21:
Amyotrophic lateral sclerosis type 1
Autoimmune polyendocrine syndrome
Autoimmune polyendocrine syndrome type 1
Holocarboxylase synthetase deficiency
Jervell and Lange-Nielsen syndrome
Majewski osteodysplastic primordial dwarfism type II(MOPD II, or MOPD2)
Chromosome 22
The following diseases are some of those related to genes on chromosome 22:
Desmoplastic small round cell tumor
22q13 deletion syndromeor Phelan-McDermid syndrome
The following conditions are caused by changes in the structure or number of copies of chromosome 22:
2 deletion syndrome: Most people with 22q11.2 deletion syndrome are missing about 3 million base pairs on one copy of chromosome 22 in each cell. The deletion occurs near the middle of the chromosome at a location designated as q11.2. This region contains about 30 genes, but many of these genes have not been well characterized. A small percentage of affected individuals have shorter deletions in the same region.
The loss of one particular gene, TBX1, is thought to be responsible for many of the characteristic features of 22q11.2 deletion syndrome, such as heart defects, an opening in the roof of the mouth (a cleft palate), distinctive facial features, and low calcium levels. A loss of this gene does not appear to cause learning disabilities, however. Other genes in the deleted region are also likely to contribute to the signs and symptoms of 22q11.2 deletion syndrome.
22q13 deletion syndrome(Phelan-McDermid syndrome): The deletion of the distal tip of the chromosome 22 is related to moderate to severe developmental delay and mental retardation. This region includes the Shank3 gene, thought to be responsible for the neurological deficits of the syndrome (Wilson et al., 2003).
Almost all children affected by the 22q13 deletion have absent or severely delayed speech; minor facial dysmorphism; thin, flaky toenails; large, fleshy hands; large feet; prominent, poorly formed ears and other characteristics which are not visually apparent: hypotonia (97%); normal to accelerated growth (95%); increased tolerance to pain (86%); seizures (unknown percentage).
Other chromosomal conditions: Other changes in the number or structure of chromosome 22 can have a variety of effects, including mental retardation, delayed development, physical abnormalities, and other medical problems. These changes include an extra piece of chromosome 22 in each cell (partial trisomy), a missing segment of the chromosome in each cell (partial monosomy), and a circular structure called ring chromosome 22 that is caused by the breakage and reattachment of both ends of the chromosome.
Cat-eye syndromeis a rare disorder most often caused by a chromosomal change called an inverted duplicated 22. A small extra chromosome is made up of genetic material from chromosome 22 that has been abnormally duplicated (copied). The extra genetic material causes the characteristic signs and symptoms of cat-eye syndrome, including an eye abnormality called ocular iris coloboma (a gap or split in the colored part of the eye), small skin tags or pits in front of the ear, heart defects, kidney problems, and, in some cases, delayed development.
A rearrangement (translocation) of genetic material between chromosomes 9 and 22 is associated with several types of blood cancer (leukemia). This chromosomal abnormality, which is commonly called the Philadelphia chromosome, is found only in cancer cells. The Philadelphia chromosome has been identified in most cases of a slowly progressing form of blood cancer called chronic myeloid leukemia, or CML. It also has been found in some cases of more rapidly progressing blood cancers (acute leukemias). The presence of the Philadelphia chromosome can help predict how the cancer will progress and provides a target for molecular therapies.
Emanuel Syndrome is a translocation of chromosomes 11 and 22. Originally known as Supernumerary der(22) Syndrome, it occurs when an individual has an extra chromosome composed of pieces of the 11th and 22nd chromosomes.
X Chromosome
Abnormalities associated with the X chromosome.
Klinefelter syndrome:
Klinefelter syndrome is caused by the presence of one or more extra copies of the X chromosome in a male’s cells. Extra genetic material from the X chromosome interferes with male sexual development, preventing the testicles from functioning normally and reducing the levels oftestosterone.
Males with Klinefelter syndrome typically have one extra copy of the X chromosome in each cell, for a total of two X chromosomes and one Y chromosome (47,XXY). It is less common for affected males to have two or three extra X chromosomes (48,XXXY or 49,XXXXY) or extra copies of both the X and Y chromosomes (48,XXYY) in each cell. The extra genetic material may lead to tall stature, learning and reading disabilities, and other medical problems. Each extra X chromosome lowers the child’sIQ by about 15 points, which means that the average IQ in Klinefelter syndrome is in general in the normal range, although below average. When additional X and/or Y chromosomes are present in 48,XXXY, 48,XXYY, or 49,XXXXY, developmental delays and cognitive difficulties can be more severe and mild intellectual disability may be present.
Klinefelter syndrome can also result from an extra X chromosome in only some of the body’s cells. These cases are called mosaic 46,XY/47,XXY.
Triple X syndrome (also called 47,XXX or trisomy X):
This syndrome results from an extra copy of the X chromosome in each of a female’s cells. Females with trisomy X have three X chromosomes, for a total of 47 chromosomes per cell. The averageIQ of females with this syndrome is 90, while the average IQ of unaffected siblings is 100 [1]. Their stature on average is taller than normal females. They are fertile and their children do not inherit the condition. [2]
Females with more than one extra copy of the X chromosome (48,XXXX syndrome or 49, XXXXX syndrome) have been identified, but these conditions are rare.
Turner syndrome:
This results when each of a female’s cells has one normal X chromosome and the other sex chromosome is missing or altered. The missing genetic material affects development and causes the features of the condition, including short stature and infertility.
About half of individuals with Turner syndrome havemonosomy X (45,X), which means each cell in a woman’s body has only one copy of the X chromosome instead of the usual two copies. Turner syndrome can also occur if one of the sex chromosomes is partially missing or rearranged rather than completely missing. Some women with Turner syndrome have a chromosomal change in only some of their cells. These cases are called Turner syndrome mosaics (45,X/46,XX).
Y Chromosome
Abnormalities associated with the Y chromosome.
Y chromosome-linked diseases can be of more common types, or very rare ones. Yet, the rare ones still have importance in understanding the function of the Y chromosome in the normal case.
More common
No vital genes reside only on the Y chromosome, since roughly half of humans (females) do not have a Y chromosome. The only well-defined human disease linked to a defect on the Y chromosome is defective testicular development (due to deletion or deleterious mutation of SRY). However, having two X chromosomes and one Y chromosome has similar effects. On the other hand, having Y chromosome polysomy has other effects than masculinization.
Y chromosome microdeletion
Y chromosome microdeletion (YCM) is a family of genetic disorders caused by missing genes in the Y chromosome. Many affected men exhibit no symptoms and lead normal lives. However, YCM is also known to be present in a significant number of men with reduced fertility or reduced sperm count.
Defective Y chromosome
This results in the person presenting a female phenotype even though that person possesses an XY karyotype (i.e., is born with female-like genitalia). The lack of the second X results in infertility. In other words, viewed from the opposite direction, the person goes through defeminization but fails to complete masculinization.
The cause can be seen as an incomplete Y chromosome: the usual karyotype in these cases is 44X, plus a fragment of Y. This usually results in defective testicular development, such that the infant may or may not have fully formed male genitalia internally or externally. The full range of ambiguity of structure may occur, especially if mosaicism is present. When the Y fragment is minimal and nonfunctional, the child is usually a girl with the features of Turner syndrome or mixed gonadal dysgenesis.
XXY
Main article: Klinefelter syndrome
Klinefelter syndrome (47, XXY) is not an aneuploidy of the Y chromosome, but a condition of having an extra X chromosome, which usually results in defective postnatal testicular function. The mechanism is not fully understood; the extra X does not seem to be due to direct interference with expression of Y genes.
XYY
Main article: XYY syndrome
47,XYY syndrome (simply known as XYY syndrome) is caused by the presence of a single extra copy of the Y chromosome in each of a male’s cells. 47, XYY males have one X chromosome and two Y chromosomes, for a total of 47 chromosomes per cell. Researchers have found that an extra copy of the Y chromosome is associated with increased stature and an increased incidence of learning problems in some boys and men, but the effects are variable, often minimal, and the vast majority do not know their karyotype. When chromosome surveys were done in the mid-1960s in British secure hospitals for the developmentally disabled, a higher than expected number of patients were found to have an extra Y chromosome. The patients were mischaracterized as aggressive and criminal, so that for a while an extra Y chromosome was believed to predispose a boy to antisocial behavior (and was dubbed the “criminal karyotype”). Subsequently, in 1968 in Scotland the only ever comprehensive nationwide chromosome survey of prisons found no overrepresentation of 47,XYY men, and later studies found 47,XYY boys and men had the same rate of criminal convictions as 46,XY boys and men of equal intelligence. Thus, the “criminal karyotype” concept is inaccurate and obsolete.
Rare
The following Y chromosome-linked diseases are rare, but notable because of their elucidating of the nature of the Y chromosome.
More than two Y chromosomes
Greater degrees of Y chromosome polysomy (having more than one extra copy of the Y chromosome in every cell, e.g., XYYY) are rare. The extra genetic material in these cases can lead to skeletal abnormalities, decreased IQ, and delayed development, but the severity features of these conditions are variable.
XX male syndrome
XX male syndrome occurs when there has been a recombination in the formation of the male gametes, causing the SRY-portion of the Y chromosome to move to the X chromosome. When such an X chromosome contributes to the child, the development will lead to a male, because of the SRY gene.
For more information, go to: http://ghr.nlm.nih.gov/handbook/basics/chromosome
All of the above information was located and can be further investigated on http://en.wikipedia.org/wiki/Chromosome
AO Scan YouTube
Connect With Us
385 208-9200
215 South Mountainlands Drive
Orem, Utah 84058 USA